Research Report
Transcriptome Sequence of Litopenaeus vannamei Based on High-throughput Sequencing 
 Author
Author  Correspondence author
Correspondence author
International Journal of Marine Science, 2014, Vol. 4, No. 62 doi: 10.5376/ijms.2014.04.0062
Received: 25 May, 2014 Accepted: 07 Jul., 2014 Published: 24 Jul., 2014
Chen X.H., Zeng D.G., Chen X.L., Xie D.X., Zhao Y.Z., Yang C.L., Ma N., and Li Y.M., 2018, Transcriptome sequence of Litopenaeus vannamei based on high-throughput sequencing, International Journal of Marine Science, 4(62): 1-7 (doi: 10.5376/ijms.2014.04.0062)
Litopenaeus vannamei shrimp is the most extensively farmed crustacean species in the world. However, the genome and transcriptome data of Litopenaeus vannamei is still relatively lacking. To obtain transcriptome information of Litopenaeus vannamei, the transcriptome of Litopenaeus vannamei hepatopancreas was sequenced by 454 high-throughput sequencing technology. A total of 500,177 ESTs were produced, with an average length of 363 bp. De novo assembly of ESTs data generated 20,225 unigenes between 50 and 8,980 bp in length, with an average length of 507 bp. Similarity searches against the NCBI-Nr database revealed that 13,676 (68%) of these unigenes have significant matches (E-value<10-5). In addition, the unigenes were similarity search against the GO, COG and KEGG databases, and were annotated with gene functional descriptions, gene ontology terms, or pathways. By high-throughput sequencing, we obtained abundant transcriptome information that could contribute to novel gene identification and genome research in Litopenaeus vannamei.
Background
Litopenaeus vannamei, also called whiteleg shrimp, is one of prawn cultivars of the largest breeding in the world (Zhou et al., 2012). It is also the main prawn cultivars in the south of China. Litopenaeus vannamei has thin shell and fat body, of which the meat is fresh and tender. The shrimp grows in a fast, uniform and homogeneous speed and it has strong disease resistance. Therefore, it is popular in markets of China and other countries (Ma et al., 2008). Based on the huge commercial value and important evolution status, more and more scholars are attracted to study the growth, development, reproduction, immunity, inherence and other fields of Litopenaeus vannamei (Liu et al., 2010). However, studies of these fields are affected because of the lack of the genome and transcriptome data of L. vannamei at present. EST of gene can be used for transcriptome study. Until now, GenBank has had 162,933 EST of L. vannamei, which can be applied to clone functional genes, dig molecular marker and design cDNA chips. But the data available now still cannot meet the research needs. Moreover, less than 10 thousand of these EST in L. vannamei have been assembled and annotated, significantly restricting the use of the EST database. There have been some research reports about transcriptomes in L. vannamei for now (Clavero-Salas et al., 2007; Robalino et al., 2007), while they are mainly conducted by conventional Sanger sequencing or cDNA chips. In conventional Sanger sequencing, cDNA library needs to be established at first, then sequencing the cloned genes, which are proved to be complicated experimental processes with high cost and long time. cDNA is a fast and high-throughput method, but the gene sequence shall be known firstly, while the few genome resources of L. vannamei restricts the application of that method.
High-throughput sequencing concurrently sequences millions of DNA molecules on a chip and it can produce a huge amount of sequence data at one time, which shows the epochal revolution of sequencing technology, therefore it is called the Next Generation Sequencing (NGS). Meanwhile, high-throughput sequencing can deeply analyze the genome or transcriptome of one species, thus, it is also called deep sequencing (Margulies et al., 2005). Next generation sequencing is divided into the second generation and the third generation sequencing technology. Sequencing along with synthesis is the core idea of second generation sequencing technology, which is to determine DNA sequence by capturing the mark on the newly composited end. The third generation sequencing technology is single-molecule sequencing based on nanopores. For now, the technology platform of second generation sequencing technology mainly includes Roche 454, Illumina Solexa and Applied Biosystems Solid (Liu et al., 2012). These three platforms have their own advantages, among which Roche 454 has longer sequencing fragment and the read length can reach 400 bp above in high quality now. Compared with Solid from ABI and Solexa from Illumina company, Roche 454 is extremely suitable for genome or transcriptome sequencing of species without genome reference sequence (You et al., 2011). In this research, sequencing technology of 454 pyrophosphoric acids was applied for high-throughput transcriptome sequencing in hepatopancreas of L. vannamei and the EST data obtained was analyzed. Through the study, over 500 thousand EST sequences in high quality were acquired and carried sequence assembly, function annotation, KEGG pathway analysis and other researches, which provided abundant resources for L. vannamei genomics and improved the understanding of L. vannamei transcriptome.
1 Results
1.1 Sequencing and sequence assembly
We used the mRNA extracted from hepatopancreas of L. vannamei to construct cDNA library and performed 454 pyrosequencing on the cDNA library. The high throughput sequencing data was submitted to NCBI sequence read archive (SRA) and the accession number was SRX181883. After removing the low-quality sequence, short sequence and adapter sequence, 500,177 EST sequences of L. vannamei were obtained and the length was from 41 bp to 620 bp with the average of 363 bp. 20,225 unigenes were obtained after assembly and the length was from 50 bp to 8,980 bp with the average of 507 bp (Figure 1).
|
Figure 1 Length distribution of unigenes Note: The X axis shows the unigene length (bp); The Y axis shows the number of the corresponding unigenes |
.png)
.png)
1.2 Gene function and annotation
Similarity search (E<10-5) was used on all Nr protein database in unigene and National Center of Biotechnology Information (NCBI). The result showed that total of 13,676 unigenes (68%) was homologous with the known genes in database, and the other 6,549 unigenes (32%) had lower homology with these known genes, indicating to be new genes.
1.3 COG function annotation and classification
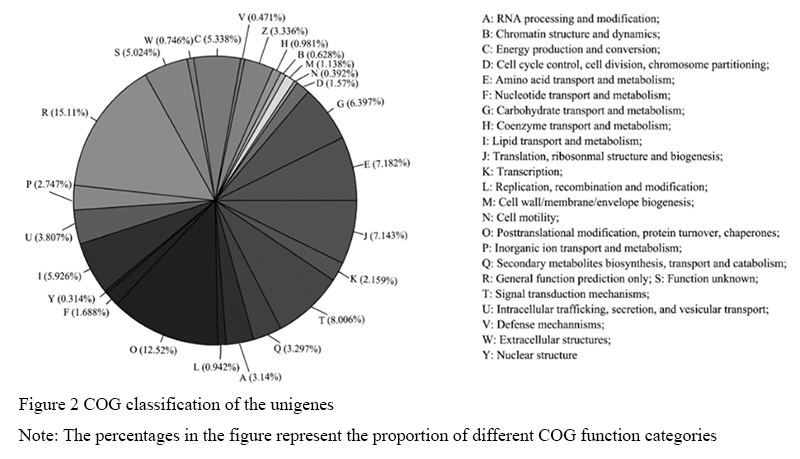
COG is a database for genetic product classification, among which every protein is assumed to come from homologous ancestral protein and the whole database is constructed based on the complete genome coding proteins in bacteria, algae and eukaryotic cells and clear phylogenetic relationships (Tatusov et al., 2000). Alignment between all unigenes and COG database demonstrated that 4,645 unigenes were annotated and divided into 25 COG categories (Figure 2). Among them, general function prediction only costed the largest percentage, followed by posttranslational modification, protein turnover, chaperones and signal transduction mechanisms. While nuclear structure, defense mechanisms, cell motility and other categories costed the least percentage.
|
Figure 2 COG classification of the unigenes Note: The percentages in the figure represent the proportion of different COG function categories |
.png)
1.4 Functional annotation and classification of GO
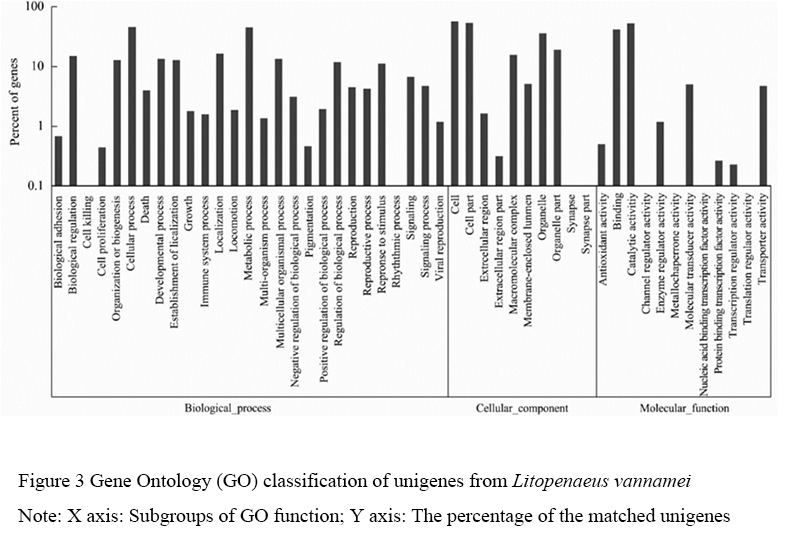
Go is a database that uses standard vocabulary for dynamic updates to describe the functions of genes and their products. It is now widely used in the study of biological transcriptome data analysis. Go is divided into 3 major functional categories, which describe the molecular function of the gene (molecular function), the location of the gene in the cell (cellular component) and the biological process the gene participated, respectively. The comparison between all unigenes and GO database showed that a total of 4,067 unigenes (20%) were annotated and categorized into 49 functional subclasses (Figure 3). The results showed that unigene of 8 functional subclasses such as cell killing, rhythmic process, synapse, channel regulator activity, metallo chaperone activity, nucleic acid binding transcription factor activity, translation regulator activity of L. vannamei was very few, while the unigene distribution of other functional subgroups was relatively balanced.
|
Figure 3 Gene Ontology (GO) classification of unigenes from Litopenaeus vannamei Note: X axis: Subgroups of GO function; Y axis: The percentage of the matched unigenes |
.png)
1.5 KEGG pathway annotation
The KEGG database is a network for recording the function and the interaction of gene products. The analysis based on the KEGG pathway will help us to further understand the biological functions of genes (Ogata et al., 1999). The analysis results showed that a total of 4,104 unigenes (20%) were annotated and classified into 176 KEGG pathways. Figure 4 shows the top 20 KEGG Pathways in the number of genes, including metabolic pathways, phagosome, focal adhesion, tight junction, adherents junction, biosynthesis of secondary metabolites, lysosome, ribosome, oxidative phosphorylation, protein digestion and absorption, tyrosine metabolism, peroxisome, metabolism of xenobiotics by cytochrome P450, amino sugar and nucleotide sugar metabolism, protein processing in endoplasmic reticulum, fatty acid metabolism, riboflavin metabolism, aminobenzoate degradation, isoquinoline alkaloid biosynthesis and RNA transport.
|
Figure 4 KEGG classification of the unigenes, showing the top 20 most abundant KEGG pathways Note: The X axis shows KEGG classification and the Y axis shows the number of the matched unigenes |
.png)
.png)
2 Discussion
Transcriptome is a collection of RNA transcripts expressed in one or more cells (Etebari et al., 2011). Transcriptome analysis can help us to study the situation and regulation of gene transcription in cells on the whole level. In the past, Sanger sequencing and cDNA chip methods were usually used in transcriptome research, but now more and more transcriptome researches use high-throughput sequencing technology. Compared with the cDNA chip method, high-throughput sequencing does not need to design probes for known sequences in advance and can sequence transcriptome of any species, as well as provide higher detection flux and lower cost and time. It is a more ideal method for the study of transcriptome. At present, some crustaceans, such as Macrobrachium rosenbergii, Eriocheir sinensis and Balanus amphitrite, have carried out transcriptome studies using high-throughput sequencing technology (De Gregoris et al., 2011; Jung et al., 2011; He et al., 2012). In this study, 454 pyrophosphate high-throughput sequencing technology was used to sequence and analyze the transcriptome of L. vannamei hepatopancreas. Total of 20,225 unigenes were obtained by splicing. The results of comparison with NCBI-Nr database showed that 6,549 unigenes had low homology with the known genes in the database, accounting for 32% of the total number, which might be unknown genes. The remaining 13,676 unigenes (68%) had high homology with the known genes in the database, of which only 5.11% were matched with the existing sequences of L. vannamei, while most of them were matched with other species. It indicated that the existing genetic sequence of L. vannamei in the database was still very small, and the unigenes we obtained greatly enriched the genetic resources of Litopenaeus vannamei in the existing database. The average length of the sequence obtained from this study was 363 bp, which was not much different from the reading length of traditional Sanger sequencing. A relatively long reading length of sequencing can effectively reduce the error of splicing and increase the length of splicing overlap groups, which is especially important for the sequencing of species that do not have a reference genome. Up till now, there has been no report on the sequencing of the whole genome of L. vannamei. We did de novo assembly on the sequencing sequence without reference to the genome, and obtained 20,225 unigenes with an average length of 507 bp. Through sampling and sequence alignment in GenBank, we found that the quality of splicing was very good and no splicing errors were existed. Our sequencing results showed that 454 sequencing was a high efficiency, low cost and high throughput transcriptome analysis method.
GO, COG and KEGG annotations are important for further understanding the function of genes. Only about 20% of unigenes we obtained is annotated to GO, COG and KEGG databases, which is mainly due to the relatively few crustacean genes included in the international public gene database at present. Many unigenes of L. vannamei obtained in this study could not search homologous gene sequences. In spite of this, the annotation results of these three databases can help us understand more about the biological characteristics of L. vannamei. Through these annotations, we can understand the molecular function of the gene, the location of the gene in the cell, the biological process involved, the metabolic pathway or signal pathway it located, and so on, which provide data for the approaching exploration of functional genes and research of related physiological functions of L. vannamei. For example, we have found that several hundred genes are related to cellular immune function, which could be used to make gene expression profiling chips in the future and could be used to detect the immune level of L. vannamei as a biochemical indicator for the breeding of disease-resistant strains of L. vannamei.
In this study, a large number of transcriptome information of L. vannamei was obtained through high-throughput sequencing, which provided valuable data for gene cloning, molecular marker discovery and genomics research of L. vannamei. In the future, comparative transcriptome studies would also be carried out, including transcriptome studies of L. vannamei at different developmental stages, different traits, virus infection and environmental factors stresses. Part of the work is now under way.
3 Materials and Methods
3.1 Materials
The experimental prawn was collected from a specific pathogen free (SPF) family of Litopenaeus vannamei from Penaeus vannamei Genetic Breeding Center in Guangxi. These prawns were 20 in total with an average weight of about 11 g. The experimental prawns were temporarily raised in 25-26°C seawater. The hepatopancreas tissue of shrimp was taken during sampling and stored in liquid nitrogen for later use.
3.2 Methods
3.2.1 RNA extraction
Trizol Qiagen was used to extract the total RNA from the hepatopancreas of Litopenaeus vannamei according to the procedures in the instructions. The concentration of RNA was determined by spectrophotometer and the integrity of RNA was detected by 1.5% (weight/volume) agarose gel electrophoresis. After RNA was extracted, mRNA was purified by PolyATtract mRNA separation system (Promega) and collected by Reasy RNA Qiagen. Then, the mRNA of the 20 samples was mixed equally for cDNA synthesis.
3.2.2 Construction and sequencing of cDNA Library
RNA Fragment reagent kit (Illumina) was used to fragment the mixed mRNA into small fragments of 300-800 bp. After the fragmentation, the fragmented product was recycled by using the Reasy RNA cleaning kit (Qqiagen). After the recycling, the random primer and MMLV were used for reverse transcription to synthesize the first strand and later the second strand was synthesized by using DNA Polymerase Ⅰ and RNase H. The processes were as follows: (1) added 2 μL 400 μmol/L N6 Radom Primer to the recycled 14 μL RNA, and bathed at 68°C for 10 min; (2) added 2 μL dNTP, 2 μL DTT, 6 μL 5 x First Strand Buffer, 2 μL MMLVRTase and 2 μL H2O into the above RNA tube, and reacted at 42°C for 30 min; (3) added 5 μL 10 x DNA Polymerase Ⅰ Buffer, 1 μL DNA Polymerase Ⅰ (10 U), 1 μL RNase H (6 U) to the above reaction solution, added water to 50 μL, and bathed at 16°C for 2 h; (4) recycled the product with MinElute DNA Cleaning kit (Qiagen) after the reaction was completed; (5) joint connection: took 15 μL of the above recycled product and added 1 μL Adptor, 2 μL 10 x DNA Ligase Buffer and 2 μL T4 DNA Ligase, bathed at 25°C for 10 min, recycled the product with MinElute DNA Cleaning kit after warm bath, and sequenced it in 454 GS FLX system (Roche).
3.2.3 Sequence assembly, function annotation, classification and path analysis
Use SeqClean program (http://compbio.dfci.harvard.edu/tgi/software) to remove the joint sequence and the low-quality sequence. Sequence assembly was carried out with iAssembler procedure (http://bioinfo.bti.cornell.edu/tool/iAssembler) (Zheng et al., 2011). The unigenes obtained through assembly were compared with databases such as NCBI Nr, SwissProt, GO, COG and KEGG (E value<10-5), respectively, and the best annotation was selected (Zhang et al., 2010). The annotation tool used for Nr, COG and SwissProt was blastx, and that for GO was blast-2GO. KEGG pathway annotation used blastx and GenMAPP 2.1 (http://www.genmapp.org/).
Authors’ contributions
ZDG was responsible for data analysis and thesis writing. CXL, XDX, ZYZ, YCL, MN, and LYM were responsible for sample processing and experiment. CXH was responsible for the overall design and guiding the research work. All authors read and approved the final manuscript.
Acknowledgements
We thank Guangxi Penaeus Vannamei Genetic Breeding Center for providing experimental animals. This research is funded by National Natural Science Foundation Project (31160531), thanks again for its support.
Clavero-Salas A., Sotelo-Mundo R.R., Gollas-Galvan T., Hernandez-Lopez J., Peregrino-Uriarte A.B., Muhlia-Almazan A., and Yepiz-Plascencia G., 2007, Transcriptome analysis of gills from the white shrimp Litopenaeus vannamei infected with white spot syndrome virus, Fish & Shellfish Immunology, 23(2): 459-472
https://doi.org/10.1016/j.fsi.2007.01.010
PMid: 17337210
De Gregoris T.B., Rupp O., Klages S., Knaust F., Bekel T., Kube M., Burgess J.G., Arnone M.I., Goesmann A., Reinhardt R., and Clare A.S., 2011, Deep sequencing of naupliar-, cyprid- and adult-specific normalised expressed sequence tag (EST) libraries of the acorn barnacle Balanus amphitrite, Biofouling, 27(4): 367-374
https://doi.org/10.1080/08927014.2011.577211
PMid: 21526438
Etebari K., Palfreyman R.W., Schlipalius D., Nielsen L.K., Glatz R.V., and Asgari S., 2011, Deep sequencing-based transcriptome analysis of Plutella xylostella larvae parasitized by Diadegma semiclausum, BMC Genomics, 12: 446
https://doi.org/10.1186/1471-2164-12-446
PMid: 21906285
PMCid: PMC3184118
He L., Wang Q., Jin X., Wang Y., Chen L., and Liu L., 2012, Transcriptome profiling of testis during sexual maturation stages in Eriocheir sinensis using illumina sequencing, PloS one, 7(3): e33735
https://doi.org/10.1371/journal.pone.0033735
PMid: 22442720
PMCid: PMC3307765
Jung H., Lyons R.E., Dinh H., Hurwood D.A., McWilliam S., and Mather P.B., 2011, Transcriptomics of a giant freshwater prawn (Macrobrachium rosenbergii): De novo assembly, annotation and marker discovery, PloS One, 6(12): e27938
https://doi.org/10.1371/journal.pone.0027938
PMid: 22174756
PMCid: PMC3234237
Liu K.F., Chiu C.H., Shiu Y.L., Cheng W., and Liu C.H., 2010, Effects of the probiotic, Bacillus subtilis E20, on the survival, development, stress tolerance, and immune status of white shrimp, Litopenaeus vannamei larvae, Fish & Shellfish Immunology, 28(5-6): 837-844
https://doi.org/10.1016/j.fsi.2010.01.012
Liu L., Li Y., Li S., Hu N., He Y., Pong R., Lin D., Lu L., and Law M., 2012, Comparison of next-generation sequencing systems, J. Biomed. Biotechnol., 2012: 251364
https://doi.org/10.1155/2012/251364
PMid: 22829749
PMCid: PMC3398667
Ma N., Chen X.H., Zeng D.G., Peng M., and Li Y.M., 2008, SNP analysis of the Cathepsin L gene in Litopenaeus vannamei, Wuhan Daxue Xuebao (Lixueban) (Journal of Wuhan University: Natural Science Edition), 54(4): 503-506
Margulies M., Egholm M., and Altman W.E., 2005, Genome sequencing in microfabricated high-density picolitre reactors, Nature, 437(7057): 376-380
https://doi.org/10.1038/nature03959
PMid: 16056220
PMCid: PMC1464427
Ogata H., Goto S., Sato K., Fujibuchi W., Bono H., and Kanehisa M., 1999, KEGG: Kyoto encyclopedia of genes and genomes, Nucleic Acids Research, 27(1): 29-34
https://doi.org/10.1093/nar/27.1.29
PMid: 9847135
PMCid: PMC148090
Robalino J., Almeida J.S., McKillen D., Colglazier J., Trent H.F., Chen Y.A., Peck M.E., Browdy C.L., Chapman R.W., Warr G.W., and Gross P.S., 2007, Insights into the immune transcriptome of the shrimp Litopenaeus vannamei: Tissue-specific expression profiles and transcriptomic responses to immune challenge, Physiological Genomics, 29(1): 44-56
https://doi.org/10.1152/physiolgenomics.00165.2006
PMid: 17148689
Tatusov R.L., Galperin M.Y., Natale D.A., and Koonin E.V., 2000, The COG database: A tool for genome-scale analysis of protein functions and evolution, Nucleic Acids Research, 28(1): 33-36
https://doi.org/10.1093/nar/28.1.33
PMid: 10592175
PMCid: PMC102395
You F.M., Huo N., Deal K.R., Gu Y.Q., Luo M.C., McGuire P.E., Dvorak J., and Anderson O.D., 2011, Annotation-based genome-wide SNP discovery in the large and complex Aegilops tauschii genome using next-generation sequencing without a reference genome sequence, BMC Genomics, 12: 59
https://doi.org/10.1186/1471-2164-12-59
PMid: 21266061
PMCid: PMC3041743
Zhang F., Guo H., Zheng H., Zhou T., Zhou Y., Wang S., Fang R., Qian W., and Chen X., 2010, Massively parallel pyrosequencing-based transcriptome analyses of small brown planthopper (Laodelphax striatellus), a vector insect transmitting rice stripe virus (RSV), BMC Genomics, 11: 303
https://doi.org/10.1186/1471-2164-11-303
PMid: 20462456
PMCid: PMC2885366
Zheng Y., Zhao L., Gao J., and Fei Z., 2011, iAssembler: A package for de novo assembly of Roche-454/Sanger transcriptome sequences, BMC Bioinformatics, 12: 453
https://doi.org/10.1186/1471-2105-12-453
PMid: 22111509
PMCid: PMC3233632
Zhou J., Fang W., Yang X., Zhou S., Hu L., Li X., Qi X., Su H., and Xie L., 2012, A nonluminescent and highly virulent Vibrio harveyi strain is associated with “bacterial white tail disease” of Litopenaeus vannamei shrimp, PloS One, 7(2): e29961
https://doi.org/10.1371/journal.pone.0029961
PMid: 22383954
PMCid: PMC3288001



. PDF(0KB)
. HTML
Associated material
. Readers' comments
Other articles by authors
. Xiaohan Chen
. Digang Zeng
. Xiuli Chen
. Daxiang Xie
. Yongzhen Zhao
. Chunling Yang
. Ning Ma
. Yongmei Li
Related articles
. High-throughput sequencing
. Litopenaeus vannamei
. Transcriptome
Tools
. Email to a friend
. Post a comment